import abtem
import ase
import matplotlib.pyplot as plt
from ase.io import write
PACBED quickstart#
This notebook demonstrates a basic simulation of a position averaged convergent beam electron diffraction (PACBED) simulation of SrTiO\(_3\).
Configuration#
We start by (optionally) setting our configuration. See documentation for details.
abtem.config.set(
{
"device": "cpu",
"fft": "fftw",
"diagnostics.task_progress": False,
"diagnostics.progress_bar": False,
}
);
Atomic model#
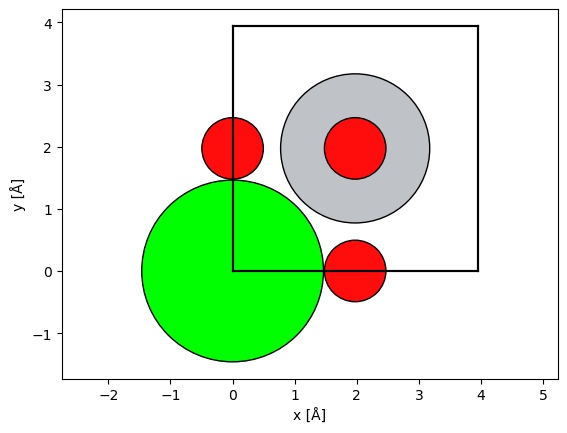
We import a model of SrTiO3. See our walkthrough or our tutorial on atomic models.
atoms = ase.io.read("data/SrTiO3.cif")
abtem.show_atoms(atoms);
Crystal potential#
potential_unit = abtem.Potential(atoms, slice_thickness=0.5, sampling=0.05)
repetitions_xy = 5
repetitions_z = 10
potential = abtem.CrystalPotential(
potential_unit, repetitions=(repetitions_xy, repetitions_xy, repetitions_z)
)
Probe#
probe = abtem.Probe(energy=100e3, semiangle_cutoff=30)
probe.grid.match(potential)
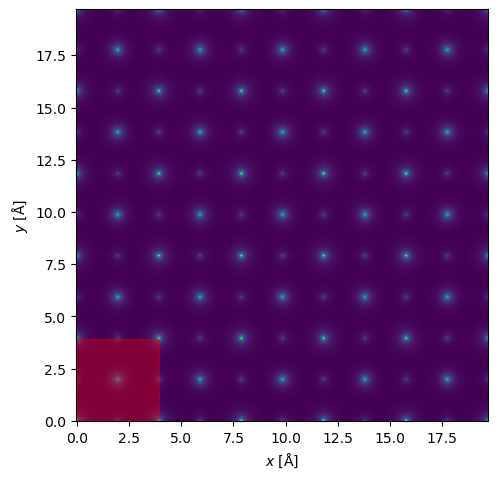
Scan#
scan = abtem.GridScan(potential=potential, start=(0, 0), end=(1 / repetitions_xy, 1 / repetitions_xy), fractional=True)
visualization = potential.show()
scan.add_to_plot(visualization)
Detect#
detector = abtem.PixelatedDetector()
measurement = probe.scan(scan=scan, potential=potential, detectors=detector)
measurement.compute();
Average over scan positions#
measurement.shape
(13, 13, 185, 185)
measurement.axes_metadata
type label coordinates
------------------- -------- --------------------
ScanAxis x [Å] 0.00 0.30 ... 3.64
ScanAxis y [Å] 0.00 0.30 ... 3.64
ReciprocalSpaceAxis kx [1/Å] -4.66 -4.61 ... 4.66
ReciprocalSpaceAxis ky [1/Å] -4.66 -4.61 ... 4.66
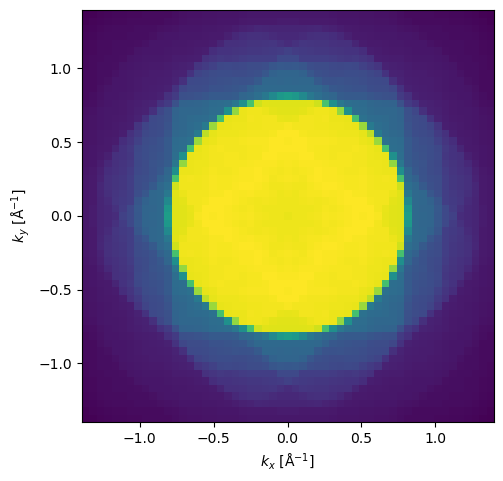
pacbed = measurement.mean(axis=(0,1))
pacbed.crop(50).show(power=0.5);
# this cell produces a thumbnail for the online documentation
visualization = pacbed.crop(50).show(power=0.5)
visualization.axis_off()
plt.savefig(
"../thumbnails/pacbed_quickstart.png", bbox_inches="tight", pad_inches=0
)
